
Fuente, 5 de diciembre de 2016
* Natalia Rojo Suárez1; *María del Rosario Marín Iglesias2; Joaquín Argente Alcaraz1; Miguel Ángel Moya Molina1
Servicio de Neurología, Hospital Universitario Puerta del Mar de Cádiz.
Servicio de Genética Médica, Hospital Universitario Puerta del Mar de Cádiz
*Ambos autores contribuyen por igual al trabajo.
Resumen
La ataxia por déficit de vitamina E (AVED) es una enfermedad rara de herencia autosómico recesiva debida a mutaciones en el gen TTPA. Se engloba entre las ataxias espinocerebelosas de origen genético con comienzo en la pubertad.
Es una entidad similar a la Ataxia de Friedreich, caracterizada por ataxia progresiva con arreflexia y pérdida de sensibilidad profunda. Casos clínicos. En el presente trabajo se va a proceder a describir dos casos familiares con clínica sugestiva, patrón de herencia recesivo, en los que la determinación de los niveles de vitamina E en sangre orientó hacia el diagnóstico de AVED.
El estudio genético en busca de mutaciones en el gen TTPA confirmó la sospecha diagnóstica en ambos casos, presentando las mutaciones c.744delA y c.513_514insTT, que son las que con más frecuencia se presentan en nuestra área. Conclusión.
Debemos sospechar que estamos ante esta patología en pacientes con fenotipo Ataxia de Friedreich con estudio genético negativo y niveles de vitamina E bajos. El análisis de la secuencia del gen TTPA en la mayoría de los casos nos permitirá no sólo confirmar la enfermedad sino también diagnosticar casos presintomáticos entre los familiares del caso índice.
Este hecho es de suma importancia, puesto que se ha demostrado que la instauración precoz de tratamiento con vitamina E puede evitar el desarrollo de la sintomatología o al menos de las complicaciones propias de la progresión de la enfermedad.
Palabras clave: Ataxia cerebelosa; AVED; Proteina Transferidora de Alfa-Tocoferol; Ataxia con déficit de Vitamina E, TTPA
Descargar artículo completo en PDF
INTRODUCCIÓN
La ataxia por déficit de vitamina E (AVED) es una enfermedad neurodegenerativa rara, descrita por primera vez por Burck et al. en 1991 y que se caracteriza por dar lugar a una ataxia progresiva debido a la pérdida de la sensibilidad propioceptiva, ausencia de reflejos y un marcado déficit de vitamina E en suero (Anheim et al., 2012). Presenta una herencia autosómico recesiva, ocasionada por una mutación en el gen que codifica para la proteína transferidora de α-tocoferol (TTPA). El AVED es la segunda ataxia cerebelosa hereditaria más prevalente en el área Mediterránea tras la ataxia de Friedreich (El Euch-Fayache et al., 2014).
La prevalencia se estima en 1/300.000 habitantes. Afecta a sujetos generalmente en la pubertad, aunque el rango de edad es variable (entre 5-20 años). Además de la ataxia, a menudo se pueden observar otras alteraciones típicas de afectación cerebelosa (adiadococinesia, disartria o dismetría) y temblor cefálico; en ocasiones se puede acompañar de alteraciones en la agudeza visual (retinosis pigmentaria), de afectación de la sensibilidad profunda propioceptiva y de la vía piramidal. Por otro lado, en una misma familia la edad de inicio y el curso clínico de la enfermedad suelen ser uniformes, variando ampliamente el fenotipo y la gravedad del cuadro si estudiamos a sujetos de diferentes familias (El Euch-Fayache et al., 2014).
Los enfermos con AVED presentan unas características clínicas muy similares a la ataxia de Friedreich, mucho más frecuente, como son la edad de inicio, la presencia de síntomas cerebelosos, la afectación piramidal, la alteración de la sensibilidad profunda, la arreflexia y las deformidades esqueléticas secundarias a largo plazo; lo que da lugar a confusiones en el diagnóstico en muchos casos. Sin embargo, existen algunas diferencias clínicas que pueden orientarnos hacia una u otra entidad (Ver tabla 1). Por ejemplo, el temblor cefálico es raramente encontrado en la ataxia de Fredreich, siendo la miocardiopatía más frecuente en esta entidad. La retinitis pigmentaria y la distonía son exclusivas de la AVED y la diabetes lo es de la ataxia de Friedreich. La edad de inicio en la AVED oscila entre los 5-20 años de edad con un fenotipo muy variable, siendo las más graves aquellas con comienzo precoz por asociarse a un mayor riesgo de desarrollar miocardiopatía (Anheim et al., 2012).
Tabla 1. Diferencias clínicas entre la ataxia de Friedreich y la ataxia por déficit de vitamina E.
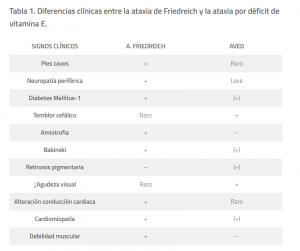
+/- → Síntoma generalmente presente/ ausente.
(+) → Síntoma presente sólo en determinadas mutaciones.
Se debe sospechar esta entidad ante un paciente con fenotipo de ataxia de Friedreich que se acompañe de una marcada disminución de la concentración de vitamina E, siempre en ausencia de causas de malabsorción digestiva, que han de ser descartadas para el diagnóstico de esta entidad. Los hallazgos de laboratorio revelan un déficit importante de vitamina E en suero, y a diferencia de lo que ocurre en los síndromes malabsortivos, los niveles de lípidos y lipoproteínas permanecen normales. Las técnicas de neuroimagen a veces no son útiles para el diagnóstico, puesto que no muestran una atrofia cerebelosa evidente u otros signos de interés en las primeras etapas de la enfermedad. La electromiografía revela generalmente una neuropatía sensorial pura. El estudio molecular del gen TTPA es capaz de detectar mutación en el 90% de los individuos que presentan alta sospecha clínica de la enfermedad.
En el presente trabajo se describen dos casos familiares de esta patología diagnosticados en nuestro hospital y que dan lugar a la discusión posteriormente expuesta.
CASOS CLÍNICOS
Caso 1
Mujer de 29 años que es derivada a consultas de Neurología por un cuadro de ataxia progresiva. Debutó con los primeros síntomas a los 10 años de edad, manifestándose con un temblor distónico cefálico y alteración en la marcha, siendo diagnosticada de forma errónea en otro centro de ataxia de Friedreich. Desde su inicio, la paciente presentó un empeoramiento progresivo e insidioso del cuadro clínico, con caídas cada vez más frecuentes hasta dar lugar a una gran inestabilidad para la marcha que la obligó a necesitar una silla de ruedas para los desplazamientos y ayuda para sus cuidados durante las 24 horas del día. Entre los antecedentes familiares, según refiere la paciente dos de sus cuatro hermanos presentaban una sintomatología similar. No existen datos de consanguinidad parental (Figura 1).
Figura 1. Árbol genealógico de la familia de la paciente del caso 1.
En el examen físico la paciente manifestaba alteración en el lenguaje, con disartria y habla escándida; el balance motor mostraba una tetraparesia leve de predominio proximal con disminución de los movimientos finos con ambas manos; los reflejos de estiramiento muscular estaban abolidos y con un reflejo cutáneo-plantar extensor bilateral; en la exploración sensitiva superficial existía abolición distal en ambos miembros inferiores y estaba disminuida en ambos miembros superiores, con apalestesia en las cuatro extremidades; la exploración cerebelosa ponía de manifiesto una dismetría troncal y de miembros con la prueba índice- nariz de forma bilateral. Los hallazgos clínicos hicieron sospechar en primera instancia, dada su mayor frecuencia en la población, una ataxia de Friedreich.
En las pruebas complementarias realizadas, los resultados de los estudios analíticos muy completos no mostraron ningún hallazgo de interés (entre ellos, metabolismo del cobre, hormonas tiroideas y el nivel de coenzima Q10 en sangre). Las pruebas de neuroimagen llevadas a cabo no revelaban alteración a nivel cerebeloso ni troncoencefálico, ni ningún otro dato a destacar. El estudio neurofisiológico realizado al inicio del cuadro no aportaba ningún dato relevante para el caso, sin haberse vuelto a realizar esta prueba a lo largo de su seguimiento. El estudio cardiológico realizado entonces no mostró hallazgos de la conducción ni alteraciones a nivel estructural. Se solicitó un estudio genético con objeto de detectar ataxia de Friedreich mediante reacción en cadena de la polimerasa y poner de manifiesto la expansión en homocigosis del trinucleótido GAA en el intrón 1 del gen FXN, presentando dos alelos dentro de la normalidad. Además se descartaron mediante pruebas genéticas las ataxias espinocerebelosas del adulto, SCA1, SCA2, SCA3, SCA6, SCA7 y DRPLA.
Posteriormente, durante el seguimiento de la paciente, se determinaron los valores de vitamina E en suero, mostrando cifras bajas para la edad de la paciente (2,3 µg/ml; val. Ref. Adultos: 5-20 µg/ml). Tras este resultado se orientó el cuadro hacia una posible ataxia espinocerebelosa por déficit de vitamina E que fue confirmado al solicitar el estudio genético y detectar la mutación c.744delA en homocigosis en el gen α-tocoferol mediante secuenciación directa de los 5 exones y de las regiones intrónicas adyacentes de dicho gen. Desde entonces, la paciente se mantiene en tratamiento con vitamina E, se encuentra estable y con un notable enlentecimiento en la progresión de su patología, si bien la clínica ya descrita no ha podido ser revertida. Los hermanos de la paciente no fueron estudiados en este hospital al ser residentes en otro país.
Caso 2
Mujer de 28 años valorada en consultas externas de Neurología, a la que acude derivada de otro especialista por presentar distonía cervical desde hacía años acompañada de temblor cefálico de inicio reciente y temblor intencional en mano derecha. Entre sus antecedentes familiares sólo destaca una hermana menor en estudio por ataxia en otro centro. No existen datos de consanguinidad parental (Figura 2).
Figura 2. Árbol genealógico de la familia de la paciente del caso 2.
En la exploración física se observa distonía cervical con lateralización hacia la derecha y componente torsional así como temblor cefálico de alta frecuencia. El balance motor no mostró alteraciones, pero los reflejos de estiramiento muscular se encontraron abolidos. No presentaba dismetrías y existía discreta alteración de la marcha, con un leve aumento de la base de sustentación. A la inspección, se observaron unos pies cavos bilaterales con retracción aquilea, lo que orientó al estudio de la ataxia de Friedreich como primera sospecha diagnóstica.
En las exploraciones complementarias realizadas los estudios analíticos no mostraron ningún hallazgo de interés (incluyendo metabolismo del cobre, estudio de hormonas tiroideas o niveles de coenzima Q10). Las pruebas de neuroimagen resultaron normales y el estudio electroneurográfico puso de manifiesto hallazgos compatibles con una polineuropatía sensitiva axonal leve. El estudio cardiológico no mostró alteraciones morfológicas ni de la conducción.
Nuestra paciente tiene una hermana de 23 años, que estaba en estudio en otro centro por inestabilidad y desequilibrio de la marcha de inicio paulatino con sensación vertiginosa acompañante. Fue valorada en el Servicio de Otorrinolaringología no detectándose alteraciones, siendo el estudio de resonancia cerebral y de ambos conductos auditivos internos normales. Le pedimos a la paciente que viniese a la próxima consulta acompañada de su hermana y en la exploración realizada a ésta destacaba una disartria con voz escándida, una hipotonía generalizada, arreflexia global y reflejo cutáneo plantar extensor derecho. La sensibilidad profunda distal en todos los miembros estaba abolida y a nivel cerebeloso se detectó una dismetría global de las cuatro extremidades con las maniobras índice-nariz y talón-rodilla afecta. Además, presentaba una marcha atáxica, con aumento de la base de sustentación y tendencia a caer hacia la derecha.
En nuestra paciente, se comenzó a estudiar la polineuropatía sensitiva axonal mediante estudio genético de los genes PMP22, MPZ, LITAF y EGR2, no detectándose mutación en sus secuencias. Posteriormente, al tratarse de dos hermanas con un cuadro de inicio similar, se sospechó la presencia de una ataxia hereditaria de carácter autosómico recesivo, por lo que se solicitó estudio genético para ataxia de Friedreich, detectándose dos alelos en el rango de la normalidad. Al igual que en el caso 1, se realizaron estudios para las ataxias espinocerebelosas del adulto más frecuentes (SCA1, SCA2, SCA3, SCA6, SCA7 y DRPLA) no detectándose alteración en el mismo. Dado que la ataxia por deficiencia de vitamina E y la ataxia de Friedreich muestran un patrón de herencia recesivo, como el mostrado en esta familia, y una edad de comienzo de la enfermedad similar, se solicitaron las concentraciones de vitamina E en suero en ambas pacientes, observándose unos niveles reducidos de los mismos sólo en la hermana más joven. Una vez confirmado dicho déficit se analizó la secuencia del gen TTPA en ambas pacientes, detectándose que eran heterocigotas compuesta para las mutaciones c.513_514insTT y c.744delA, ambas asociadas previamente con dicha enfermedad. En nuestra paciente, se comenzó el tratamiento con vitamina E a altas dosis desde la detección del déficit, y en la actualidad se encuentra estable y mantiene una vida normal e independiente. Su hermana menor, continúa el seguimiento en su centro de referencia por lo que no disponemos de datos evolutivos al respecto.
DISCUSIÓN
La vitamina E es una vitamina liposoluble con función antioxidante que está presente en vegetales, nueces, productos lácteos, carnes y pescados, siendo esencial para el mantenimiento de una estructura y función neurológica normales. La familia de la vitamina E está formada por cuatro tocoferoles y cuatro tocotrienoles, siendo el α-tocoferol la forma que predomina en los tejidos humanos (Jiang, 2014; Ulatowski et al., 2013).
El déficit de vitamina E da lugar a anomalías neurológicas, especialmente a una ataxia espinocerebelosa y su déficit grave puede en último término ocasionar la muerte del sujeto, debido a la atrofia celular secundaria que se produce por aumento del estrés oxidativo, así como por la disminución de ramificaciones dentríticas en las neuronas de Purkinje, predominante en la corteza cerebelosa (Ulatowski et al., 2013).
La AVED, heredada con carácter autosómico recesivo, es debida a mutaciones en el gen TTPA que codifica para la proteína transferidora de a- tocoferol, mapeada en 8q12.3. Se han descrito más de 20 mutaciones, siendo la c.744delA seguida de la c.513_514insTT las más frecuentes en el área Mediterránea (Elkamil et al., 2015). En nuestros casos, se detectó la mutación c.744delA al menos en uno de los alelos; esta mutación se ha asociado con un fenotipo de inicio temprano, curso clínico rápidamente progresivo y riesgo incrementado de miocardiopatía (El Euch-Fayache et al., 2014; Bromley et al, 2013).
En la AVED la absorción intestinal de vitamina E debe ser normal pero los niveles encontrados son bajos debido a que las mutaciones en el gen TTPA provocan una defectuosa incorporación hepática del α-tocoferol a las lipoproteinas de baja densidad (VLDLs). En condiciones normales, la vitamina E circula por el torrente sanguíneo ligada a estas lipoproteínas. En los pacientes en los que se detecta la AVED, al aportar la vitamina E de forma terapéutica a altas dosis aumentamos su concentración a nivel hepática y al tratarse de un mecanismo concentración dependiente favorecemos el paso de tocoferol a las lipoproteínas que circulan por el torrente sanguíneo. Los niveles de vitamina E sérica por debajo de 2,5 mg/ml asociados a unos niveles normales de vitamina A son la principal característica de la AVED, a diferencia de lo observado en la abetaliproteinemia, en la cual los niveles de vitamina A son también bajos. Debido a este hallazgo, se ha sugerido que en pacientes con ataxia cerebelosa recesiva sea prioritario realizarse un rastreo de los valores de ambas vitaminas en suero, ya que su aportación exógena es imprescindible para evitar el progresivo empeoramiento de estos pacientes (Ulatowski et al, 2013; Niki y Traber, 2012).
Las técnicas de neuroimagen, en especial la resonancia magnética nuclear cerebral, pueden mostrar en la mitad de los sujetos afectos, al igual que en otras ataxias recesivas, una atrofia cerebelosa y de la médula espinal cervical que se evidencian habitualmente en etapas ya avanzadas de la enfermedad, por lo que no son pruebas útiles como método de rastreo.
A diferencia de lo que ocurre con otros tipos de ataxias hereditarias y con un gran interés de cara al pronóstico del paciente, la AVED tiene tratamiento disponible que consiste en suplementos de vitamina E a altas dosis (800-1500mg o 40mg/kg en niños). Algunos síntomas pueden ser revertidos si el tratamiento es iniciado en etapas precoces o iniciales de la enfermedad, como ocurre con otras ataxias tratables (Refsum, déficit de coenzima Q y la xantocromatosis cerebrotendinosa). En sujetos de mayor edad, la progresión de la enfermedad puede ser frenada aunque la alteración de la sensibilidad propioceptiva ya presentada así como la marcha inestable no se verán modificadas. Por otro lado, recalcar que si el tratamiento con suplementos de vitamina E es iniciado en sujetos presintomáticos portadores de las dos mutaciones causantes de la enfermedad, ni siquiera de desarrollarán los síntomas de la AVED, permaneciendo estos sujetos completamente asintomáticos el resto de su vida (El Euch-Fayache et al., 2014; Anheim et al., 2012).
Por tanto, determinar los niveles séricos de vitamina E debe de ser una prioridad médica a llevar a cabo entre todos los familiares a riesgo. De resultar la concentración anormalmente baja, debe priorizarse la búsqueda de mutaciones en el gen TTPA. Por otro lado, de resultar dentro de los parámetros normales (valores de referencia en adultos: 5-20 µg/ml), debe prestarse entonces más atención al gen FXN de la ataxia de Friedreich (FRDA). Sin embargo, esto no exime que pudiera tratarse de una mutación en el gen TTPA, como lo atestigua la presencia de niveles normales de vitamina E en una de las pacientes del caso 2. Es importante destacar en estos casos la posible presencia de falsos negativos dada la baja sensibilidad en la determinación de los niveles séricos de vitamina E.
Las mutaciones en el gen TTPA pueden buscarse secuenciando el propio gen directamente o bien merced paneles de secuenciación masiva de next generation sequencing (NGS), con la ventaja de desvelarse en este caso mutaciones puntuales en otros genes relacionados con ataxia incluidos en el panel como el propio FXN de la FRDA, donde encontraremos sus mutaciones puntuales, que son de baja prevalencia pero no inexistentes, y por lo tanto, deberán ser tenidas en cuenta.
Como conclusión, la AVED es una entidad hereditaria que debemos sospechar en aquellos casos de ataxia con un patrón de herencia autosómico recesivo en los que se haya descartado por su frecuencia la ataxia de Friedreich. La detección precoz de esta entidad y el tratamiento temprano con suplementos de vitamina E a dosis altas y mantenido de por vida evitarán su progresión, que de lo contrario llevaría al sujeto hacia un estado de gran dependencia en la que ya sólo son útiles medidas con fines paliativos. La vital importancia del estudio genético radica no sólo en la detección de estos paciente, sino principalmente en la detección de casos presintomáticos dentro de una misma familia, favoreciendo el tratamiento de forma temprana y evitando así la aparición de esta grave enfermedad.
REFERENCIAS
Anheim M, et al. The Autosomal Recessive Cerebellar Ataxias. N Engl J Med. 2012; 366: 636-646. doi:10.1056/NEJMra1006610.
Bromley D, et al. Structural consequences of mutations to the α-tocopherol transfer protein associated with the neurodegenerative disease ataxia with vitamin E deficiency. Biochemistry. 2013; 52(24): 4264-4273. doi: 10.1021/bi4001084.
El Euch-Fayache G, et al. Molecular, clinical and peripheral neuropathy study of Tunisian patients with ataxia with vitamin E deficiency. Brain. 2014; 137(Pt 2):402-410. doi: 10.1093/brain/awt339.
Elkamil A, et al. Ataxia with vitamin E deficiency in Norway. J Mov Disord. 2015; 8(1):33-36. doi: 10.14802/jmd.14030.
Gene Reviews. URL: http://www.ncbi.nlm.nih.gov/books/NBK1241/[10.03.2015].
Jiang Q. Natural forms of vitamin E: metabolism, antioxidant, and anti-inflammatory activities and their role in disease prevention and therapy. Free Radic Biol Med. 2014; 72: 76-90. doi: 10.1016/j.freeradbiomed.2014.03.035.
Niki E, Traber MG. A history of vitamin E. Ann Nutr Metab. 2012; 61(3): 207-212. doi: 10.1159/000343106.
Ulatowski L, et al. Vitamin E is essential for Purkinje neuron integrity. Neuroscience. 2014; 260: 120-129. doi: 10.1016/j.neuroscience.2013.12.001.








Hola necesito presupuesto para realizarme estudio de genética ataxia.
¡Buenas tardes!
Nosotros tan solo somos una federación de personas relacionadas con la ataxia.
No estamos capacitados para emitir o gestionar este tipo de exámenes.
Deberías comentarlo con tus profesionales de la salud, y ellos en caso de considerarlo necesario te derivarán al especialista encargado de realizar ese tipo de exámenes considerando tu historial médico.
Un saludo, esperamos haber resultado de ayuda.